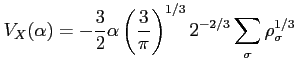Jacek Kobus
2 Dimensional Finite Difference
Hartree-Fock Program
User's Guide
ver. 2.0 (June 2010)
An input data file consists of separate lines each containing
- -
- a label
- -
- a label followed by a string of characters, integer(s)
and/or real number(s)
- -
- a string of characters, integer(s) and/or real number(s)
Real numbers can be written in a fix point or scientific notation.
Note that
- -
- labels and strings can be in upper or lower case,
- -
- the compulsory labels must follow the order given below; the
optional ones can be inserted anywhere between the title
and stop labels,
- -
- optional parameters are enclosed in square brackets,
- -
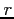 denotes a real number,
denotes a real number, 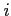 - an integer,
- an integer, 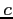 - a string
of characters,
- a string
of characters,
- -
- an exclamation mark or a hash placed anywhere in an input
line starts a comment and what follows ``!'' or ``#'' is ignored.
The following labels must be specified in the given order:
- TITLE
- Format:
- title
is any
string of up to 74 characters describing the current case. This
string is added as a header to a text file with extension dat that
contains basic data identifying a given case, i.e. atomic numbers of
nuclei, grid size and the number of electrons and orbital and exchange
functions.
- NUCLEI
- Format:
- nuclei
![$ Z_A \;\;Z_B\;\; R \;\;[\; c\;]$](img4.png)
Set the nuclei charges and the bond length.
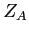 :
:- nuclear charge of centre A (real)
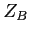 :
:- nuclear charge of centre B (real)
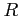 :
:- bond length (real)
-
:
 - the internuclear separation can be given in
angstrom units if this string is included (the conversion factor
0.529177249 is used)
- the internuclear separation can be given in
angstrom units if this string is included (the conversion factor
0.529177249 is used)
If
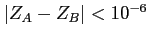 then the molecule is considered to be a
homonuclear one (this threshold can be changed by redefining
HOMOLEVL variable in blk_data).
then the molecule is considered to be a
homonuclear one (this threshold can be changed by redefining
HOMOLEVL variable in blk_data).
- CONFIG
- Format:
- config
-
:
- the total charge of a system
The following cards define molecular orbitals and their occupation.
Note that the last orbital description card must contain the
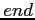 label.
label.
The possible formats are:
- Format:

-
:
- number of fully occupied orbitals of a given irreducible
representation (irrep) of the
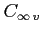 group;
two electrons make
group;
two electrons make 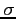 orbitals fully occupied
and four electrons are needed for filling the orbitals of other symmetries
orbitals fully occupied
and four electrons are needed for filling the orbitals of other symmetries
-
:
- symbol of the
irrep to which the orbitals
belong (sigma, pi, delta or phi)
- Format:
-
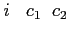
-
:
- number of fully occupied orbitals of a given irrep of the
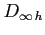 group
group
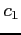 :
:- symbol of the
irrep to which the orbitals belong
(sigma, pi, delta or phi)
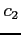 :
:- symbol for the inversion symmetry of the
irreps
(u or g)
Use this format for a homonuclear molecule unless break card
is included (see below).
- Format:
-
![$ i\;\;\;c_1\;\;c_2\;\;[c_3\;\;[\;c_4\;\;[\;c_5\;]\;]\;]$](img18.png)
-
:
- number of orbitals of a given irrep of the
group
-
:
- symbol for the
irreps to which the
orbitals belong (sigma, pi, delta, phi)
-
-
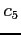 :
:
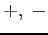 or . (a dot);
or . (a dot); 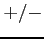 denotes spin up/down
electron and . denotes an unoccupied spin orbital
denotes spin up/down
electron and . denotes an unoccupied spin orbital
- Format:
-
![$ i\;\;\;c_1\;\;c_2\;\;c_3\;\;[\;c_4\;\;[\;c_5\;\;[\;c_6\;]\;]\;]$](img22.png)
-
:
- number of orbitals of a given irrep of the
group
-
:
- symbol for the
irrep to which the
orbitals belong (sigma, pi, delta, phi)
-
:
- symbol for the inversion symmetry of the
irrep (u or g)
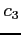 -
-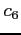 :
:-
or . (a dot);
denotes spin up/down
electron and . denotes an unoccupied spin orbital
- GRID
Two possible formats are (the second one is deprecated and is retained
for backward compatibility):
- Format:
- grid
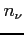
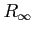
An integer and a real define a single two-dimensional grid.
-
:
- the number of grid points in
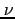 variable
variable
-
:
- the practical infinity
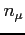 is calculated so as to make the step size in
is calculated so as to make the step size in 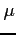 variable
equal to the stepsize in
variable.
and
have to
meet special conditions. If the conditions are not fulfilled the
nearest (but smaller) appropriate values are used.
variable
equal to the stepsize in
variable.
and
have to
meet special conditions. If the conditions are not fulfilled the
nearest (but smaller) appropriate values are used.
- Format:
- grid
Two integers and one real define a single two-dimensional grid.
-
:
- the number of grid points in
variable
-
:
- the number of grid points in
variable
-
:
- the practical infinity
and
have to meet special conditions. If the
conditions are not fulfilled the nearest (but smaller) appropriate
values are used.
- ORBPOT
- Format:
- orbpot
where
a character string determining the initial source of
orbitals and potentials. Its allowed values are:
- STOP
- Format:
- stop
This label indicates the end of input data.
The following additional labels can be specified in any order:
- BREAK
- Format:
- break
When this label is present homonuclear
molecules are calculated in
symmetry and the
symmetry labels (u or g) are superfluous.
- CONV
- Format:
- conv
![$ [\;i_1\;[\;i_2\;[\;i_3\;]\;]\;]$](img42.png)
Sometimes the convergence
threshold (for energy and/or normalization) is set too low and
cannot be satisfied on a given grid and as a result the SCF/SOR
process continues in vain. The iterations are stopped if orbital
energies or orbital norms display no improvment over a given number
of 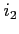 and
and 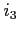 most recent iterations, respectively (the default
values are set to 20). The monitoring begins after
most recent iterations, respectively (the default
values are set to 20). The monitoring begins after 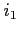 initial
iterations (default 600).
initial
iterations (default 600).
- DEBUG
- Format:
- debug
![$ [\; i_1 \; [\;i_2 \ldots \;] \;i_{40}\;]$](img46.png)
Up to 40 different debug flags can be set at a time2. If the integer 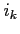 is encountered the debug
flag
is set, i.e. idbg
is encountered the debug
flag
is set, i.e. idbg
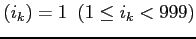 . These
are used to generate additional printouts by adding where needed the
lines of the form
. These
are used to generate additional printouts by adding where needed the
lines of the form
if (idbg(ik).eq.1) then
print *, ``debuging something ...''
...
endif
These are used to generate debug information.
- DFT
- Format:
- dft
![$ [\;c_1\;]\;\; [\;c_2\;]$](img49.png)
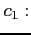
- specifies the type of DFT exchange potential to be used in
Fock equations
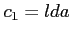 - the local density approximation with the potential
- the local density approximation with the potential
where 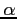 is by default set to 2/3 (the Slater exchange
potential). To change this value use the xalpha label.
is by default set to 2/3 (the Slater exchange
potential). To change this value use the xalpha label.
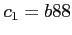 - the Becke exchange potential
- the Becke exchange potential
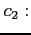
- selects the type of correlation potential to be used in
Fock equations
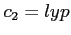 - the correlation potential of Lee, Yang and Parr
- the correlation potential of Lee, Yang and Parr
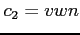 - the correlation potential of VWN
- the correlation potential of VWN
When the bare label is present and the method selected is HF then the
exchange contributions (LDA, B88, PW86 and PW91) and the correlation
contributions (LYP and VWN) to the total energy are calculated upon
completion of the SCF iterations.
- EXCHIO
- Format:
- exchio
This is a replacement for
parameter of INITIAL card (especially when
ORBPOT card is used to define the source of orbitals and
potentials). These parameters specify
how exchange potentials are to be read/written and manipulated
(stored in core memory). The program always keeps all orbitals and
Coulomb potentials in the memory. If computer resources are adequate
all exchange potentials can also be all kept there. However, during
the relaxation of a particular orbital only a fraction of exchange
potentials is needed. Thus all exchange potentials can be kept on
disk as separate files (named fort.31, fort.32,  during a
run) and only relevant ones are being retrieved when
necessary.
during a
run) and only relevant ones are being retrieved when
necessary.
The possible values of
and
are in-one,
in-many, out-one and out-many. Possible
combinations are
- FEFIELD
- Format:
- fefield
-
:
- a strength of an external static electric field directed along
the internuclear axis (in atomic units)
- FERMI
- Format:
- fermi
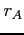
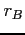
When this label is
present, the Fermi nuclear charge distribution is used. Optional
parameters
and
define the atomic masses (in amu) of
nuclei A and B. If omitted the corresponding values are taken from
the table of atomic masses compiled by Wapstra and Audi (see
blk_data).
- FIXORB
- Format:
- fixorb
This label is used to specify orbitals to be kept frozen during SCF/SOR process.
,
,
are the numbers of these orbitals as they appear on the program's
listing, i.e. their order is reversed to that used when defining the electronic
configuration (see the config card). Up to 40 different orbitals can be set at
a time. Use the bare label to keep all orbitals frozen but allow for the relaxation of
potentials.
- FIXCOUL
- Format:
- fixcoul
If this label is present then all
Coulomb potentials are kept frozen during the SCF/SOR process.
- FIXEXCH
- Format:
- fixexch
If this label is present then all
exchange potentials are kept frozen during the SCF/SOR process.
- GAUSS
- Format:
- gauss
When this label is
present, the Gauss nuclear charge distribution is used. Optional
parameters
and
define the atomic masses (in amu) of
nuclei A and B. If omitted the corresponding values are taken from
the table of atomic masses compiled by Wapstra and Audi (see blk_data).
- HOMO
- Format:
- homo
This label is used to impose explicitly
symmetry upon orbitals
of homonuclear molecules in order to improve SCF/SOR convergence.
- INOUT
- Format:
- inout
The x2dhf program can be
compiled to support calculation using three different combinations
of integer/real data types: i32 (4-byte integers, 8-byte reals), i64
(8-byte integers, 8-byte reals) and r128 (8-byte integers, 16-byte
reals); see src/Makefile for details. Strings
and
determine the combination appropriate for the format of input and
output data, respectively, and each string can be i32, i64 or r128.
In order to facilitate exchange of binary data generated on machines of different
architectures or using different compilers additional formats are available, namely
i32f, i64f or r128f which allow to export/import data in formatted instead of
unformatted form.
- INTERP
- Format:
- interp
Use this label to change the grid
between separate runs of the program. The restriction is that only
a number of grid points in one of the variables or
can
be changed at a time.
- LCAO
- Format:
- lcao
![$ [\;i\;]$](img65.png)
If the source of orbitals is
declared as hydrogen then this card must be present. In
such a case the initialization of each of the orbitals has to be
defined in terms of the linear combination of atom centred
hydrogen-like functions. For each orbital include a card of the
following format (make sure that the order of orbitals should match
the order specified under the config label):
- Format:
-
![$ c_A\;\;n_A \;\;l_A \;\;\zeta_A \;\;\;\;c_B\;\;n_B\;\;l_B\;\;
\zeta_B\;\;\;\;i_1\;\;\;\;[\;i_2\;]$](img66.png)
where
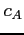 - relative mixing coefficient for a hydrogenic orbital on the
centre (real),
- relative mixing coefficient for a hydrogenic orbital on the
centre (real),
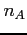 - its principle quantum number (integer)
- its principle quantum number (integer)
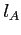 - its orbital quantum number (integer)
- its orbital quantum number (integer)
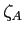 - the effective nuclear charge if
- the effective nuclear charge if 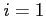 (default) or
a screening parameter if
(default) or
a screening parameter if 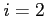 (real)
(real)
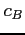 - relative mixing coefficient for a hydrogenic orbital on the
centre (real),
- relative mixing coefficient for a hydrogenic orbital on the
centre (real),
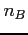 - its principle quantum number (integer)
- its principle quantum number (integer)
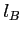 - its orbital quantum number (integer)
- its orbital quantum number (integer)
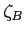 - the effective nuclear charge if
(default) or
a screening parameter if
(real)
- the effective nuclear charge if
(default) or
a screening parameter if
(real)
-
- set to 1 to freeze the orbital during scf; otherwise
set to 0 (integer)
-
- a number of successive over-relaxations for a given orbital
(integer); if omitted is set to 10
The mixing coefficients are normalized so that
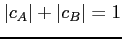
- MCSOR
- Format:
- mcsor
![$ [\;i_1\;[\;i_2\;]\;]$](img78.png)
Selects the MCSOR method for solving the Poisson equations for orbitals and potentials
(default) and changes the value of the MCSOR relaxation sweeps during a single SCF
cycle for orbitals (
) and potentials (
); by default
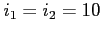 .
.
- METHOD
- Format:
- method
Select the type of calculation.
-
:
- HF - the Hartree-Fock method
-
:
- DFT - the Hartree-Fock method with the
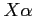 exchange
potential (
exchange
potential (
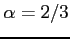 ); see the dft label to choose
another exchange or correlation potential
); see the dft label to choose
another exchange or correlation potential
-
:
- HFS - the Hartree-Fock-Slater method (Hartree-Fock with the
exchange potential) with an optimum value of the
parameter (see blk-data.inc for details)
-
:
- OED - One Electron Diatomic ground and excited states
can be calculated for the Coulomb potential in the prolate
spheroidal coordinates (default). It is also possible to specify
the Coulomb and Krammers-Henneberger potentials in cylindrical
coordinates (see the poth3 and potkh labels,
respectively). When more than one orbital is specified calculations
are carried out as if in case of a multielectron
system.3
-
:
- SCMC - the Hartree-Fock method with
exchange
where the
parameter is calculated according to the
Self-Consistent Multiplicative Constant method4
- MULTIPOL
- Format:
- multipol
[
]
-
:
- if
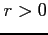 multipole moment expansion coefficients are recalculated when the
maximum error in orbital energy is changed by
(the default value is 1.15; see
setDefaults). When the maximum error is less than
the coefficients are
recalculated at least every
SCF iterations (see SCF label). To suppress
recalculation of the coefficients set
to a negative real number. This is useful when
generating potentials from a set of fixed orbitals, e.g from GAUSSIAN orbitals.
multipole moment expansion coefficients are recalculated when the
maximum error in orbital energy is changed by
(the default value is 1.15; see
setDefaults). When the maximum error is less than
the coefficients are
recalculated at least every
SCF iterations (see SCF label). To suppress
recalculation of the coefficients set
to a negative real number. This is useful when
generating potentials from a set of fixed orbitals, e.g from GAUSSIAN orbitals.
-
:
- number of terms in the multipole expansion used to calculate
boundary value for potentials (
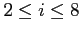 and the default is 4)
and the default is 4)
- OMEGA
- Format:
- omega
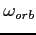 [
[
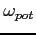 ]
]
One or two real numbers
setting over-relaxation parameters for relaxation of orbitals and potentials. The
negative value of a given parameter indicates that its value should be set to a near
optimum value obtained from a semiempirical formula (see initCBlocks and
setomega for details). If this card is omitted
is estimated from the
semiempirical formula and
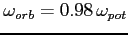 (see setDefaults
to change the default value of the scaling factor).
(see setDefaults
to change the default value of the scaling factor).
For backward compatibility the following format is also supported:
- Format:
- omega
- OMEGAOPT
- Format:
- omegaopt [ i [
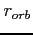 [
[ 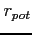 ] ] ]
] ] ]
Optional integer
parameter can be set to 1 (default) or 2. In the former case rather conservative (safe)
omega values are chosen (this is equivalent to using the omega card with the negative
values of the omega parameters). In the latter case somewhat better omega values are
chosen but faster convergence is to be expected when good initial estimates of orbitals
and potentials are available or when calculations with fixed orbitals or potentials are
performed. The near-optimal omega values obtained from a semi-empirical formula are
scaled down to produce final values used by the program. The default values of the
scaling factors for the orbital and potential overrelaxation parameters (0.986 and
0.997, respectively) can be changed by setting
and
to their desired
values.
- ORDER
- Format:
- order
An integer defining the
ordering of mesh points: 1 - natural column-wise ordering, 2 -
'middle' type of sweep (default), 3 - natural row-wise, 4 -
reversed natural column-wise (see mesh routine for details)
- POTGSZ
- Format:
- potgsz
When the OED method is chosen then this label selects a
model potential due to Green, Sellin and Zachor. For a given atom this
potential produces HF-like orbitals but it was found useful in finding decent starting
orbitals for any molecular system.
- POTGSZG
- Format:
- potgszg
When the OED method is chosen then this label selects a
model potential due to Green, Sellin and Zachor and the Gauss nuclear
charge distribution. For a given atom this potential produces HF-like orbitals
but it was found useful in finding decent starting orbitals for any molecular system.
- POTH3
- Format:
- poth3
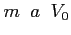
When the OED method is chosen then this
label selects a two-dimensional model potential of the form
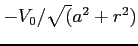 (see
kh.c for details). The following parameters can be set
(see
kh.c for details). The following parameters can be set
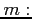
- magnatic quantum number of a state (integer)
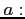
- width of the model potential (real)
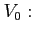
- depth of the model potential (real)
In order to get the hydrogen Coulomb potential set 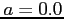 and
and
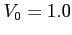 . Set
. Set 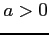 and
and 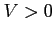 to choose its smoothed variant.
to choose its smoothed variant.
- POTKH
- Format:
- potkh
![$ m\;\;\varepsilon\;\;\omega\;\;\;[\;a\;[\;V_0\;[\;N\;]\;]\;]$](img98.png)
When the OED method is
chosen then this label selects the Krammers-Henneberger potential (see routine kh.c for
details). The following parameters can be set
-
- magnatic quantum number of a state being calculated
-
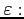
- laser intensity
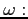
- laser cycle frequency
-
- original (before averaging over one laser cycle)
soft-core potential width (a positive real number, by default
 )
)
-
- original soft-core potential depth (by default
)
- number of intervals in the Simpson quadrature (an
integer,
 by default)
by default)
- PRINT
- Format:
- print
Up to 40 different printing flags can be set at a time. If the
integer
is encountered the printing flag
is set,
i.e. iprint
. These are used
to generate additional printouts by adding where needed the lines
of the form
if (iprint(ik).eq.1) then
print *, ``printing something ...''
...
endif
Set
See inputData routine for a list of used flags.
- PRTEVERY
- Format:
- prtevery
Subroutine pmtx can be
used to output two-dimensional arrays in a tabular row-wise form with
every
-th row and
-th column being printed (by default
every 10th row and column is selected)
- SCF
- Format:
- scf
![$ [\;i_1\;[\;i_2\;[\;i_3\;[\;i_4\;[i_5]\;]\;]\;]\;]$](img108.png)
-
:
- maximum number of scf iterations (default 1000);
to skip the scf step set
to a negative
integer,
-
:
- every
scf iterations orbitals and potentials are
saved on disk (default 20).
If
 functions are saved on disk upon completion of the scf
process. If
functions are saved on disk upon completion of the scf
process. If  functions are never written to disk,
functions are never written to disk,
-
:
- if the maximum error in orbital energy is less than
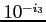 than the scf process is terminated (the
default value is 10),
than the scf process is terminated (the
default value is 10),
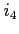 :
:- if the maximum error in orbital norm is less than
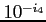 than scf process is terminated (the default
is 10),
than scf process is terminated (the default
is 10),
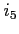 :
:- the level of output during scf process
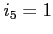 - the orbital energy, the difference between its
current and previous value, the normalization error and the
(absolute) value of the largest overlap integral between the current
orbital and all the lower lying ones of the same symmetry (the value
is zero for the lowest orbitals of each symmetry)
is printed for every orbital in every scf iteration
- the orbital energy, the difference between its
current and previous value, the normalization error and the
(absolute) value of the largest overlap integral between the current
orbital and all the lower lying ones of the same symmetry (the value
is zero for the lowest orbitals of each symmetry)
is printed for every orbital in every scf iteration
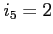 - the orbital energy, the difference between its
current and previous value and the normalization error is printed
for the worst converged orbital in energy (first line) and norm
(second line) in every scf iteration (default)
- the orbital energy, the difference between its
current and previous value and the normalization error is printed
for the worst converged orbital in energy (first line) and norm
(second line) in every scf iteration (default)
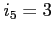 - the orbital energy, the difference between its
current and previous value and the normalization error is printed
for the worst converged orbital in energy (first line) and norm
(second line) every
iterations. Printing of ``... multipole
moment expansion coefficients (re)calculated ...'' communique is
suppressed
- the orbital energy, the difference between its
current and previous value and the normalization error is printed
for the worst converged orbital in energy (first line) and norm
(second line) every
iterations. Printing of ``... multipole
moment expansion coefficients (re)calculated ...'' communique is
suppressed
Total energy is printed every
iterations.
- SOR
- Format:
- sor
Selects the SOR method for solving the Poisson equations for orbitals and potentials
(default) and changes the value of the SOR relaxation sweeps during a single SCF
cycle for orbitals (
) and potentials (
); by default
.
- XALPHA
- Format:
- xalpha
This label allows to change
the
parameter of the LDA potential (see the HFS method and
dft label); 2/3 is its default value.
The following labels have been replaced by others but are supported for backward
compatibility with the previous version of the input data:
- INITIAL (deprecated, use ORBPOT and LCAO instead)
- Format:
- initial
![$ i_1\;[\;i_2\;[\;i_3\;]\;]$](img117.png)
-
:
- determine the initial source of orbitals and potentials:
-
:
- specifies how exchange potentials are to be read/written
and manipulated (stored in memory). The program always keeps all
orbitals and Coulomb potentials in memory. If computer resources are
adequate all exchange potentials can also be all kept in core
memory. However, during a relaxation of a particular orbital only a
fraction of exchange potentials is in fact needed. Thus all exchange
potentials can be kept on disk as separate files (named fort.31,
fort.32, ... during a run) and only relevant ones are being
retrieved when necessary.5
-
- read exchange potentials as separate files and
write them back as separate files
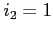 - read all exchange potentials in a file but write
them out as separate files
- read all exchange potentials in a file but write
them out as separate files
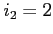 - read all exchange potentials separately but write
them out as a single file
- read all exchange potentials separately but write
them out as a single file
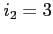 - read and write exchange potentials in the form of
a single file (default)
- read and write exchange potentials in the form of
a single file (default)
-
:
- if
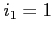 then this parameter must be set to 1 or 2 (if
omitted it is set to 1). In such a case the initialization of
each of the orbitals has to be defined in terms of the linear
combination of atom centered hydrogen-like functions
For each orbital include a card of the
following format (make sure that the order of orbitals should match
the order specified under the config label):
then this parameter must be set to 1 or 2 (if
omitted it is set to 1). In such a case the initialization of
each of the orbitals has to be defined in terms of the linear
combination of atom centered hydrogen-like functions
For each orbital include a card of the
following format (make sure that the order of orbitals should match
the order specified under the config label):
- Format:
-
where
-
- relative mixing coefficient for a hydrogenic orbital on the
centre (real),
-
- its principle quantum number (integer)
-
- its orbital quantum number (integer)
-
- the effective nuclear charge if
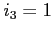 or
a screening parameter if
or
a screening parameter if 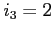 (real)
(real)
-
- relative mixing coefficient for a hydrogenic orbital on the
centre (real),
-
- its principle quantum number (integer)
-
- its orbital quantum number (integer)
-
- the effective nuclear charge if
or
a screening parameter if
(real)
-
- set to 1 to freeze the orbital during scf; otherwise
set to 0 (integer)
-
- a number of successive over-relaxations for a given orbital
(integer); if omitted is set to 10
For other values of
than 1 the orbital cards can be omitted but
then the
parameter must be set to 0.
- FIX (deprecated, use FIXORB, FIXCOUL or
FIXEXCH instead)
- Format:
- fix
If
,
or
are set to 1 then orbitals, Coulomb
potentials or exchange potentials, respectively, are kept frozen during the
scf/sor process (the respective default values are 0, 0 and 2).
If
then exchange potentials are relaxed only once during an
scf cycle.
and
cannot be set to 1 if hydrogenic
orbitals are used to initiate the orbitals.
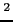 S ground state of the Th
S ground state of the Th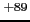 one-electron system (see
examples/oed/th+89/th+89_1s.lst).
one-electron system (see
examples/oed/th+89/th+89_1s.lst).
../examples/oed/th+89/th+89_1s.data
- First excited
S state of the Th
one-electron
system (see examples/oed/th+89/th+89_2s.lst).
../examples/oed/th+89/th+89_2s.data
- Hartree-Fock ground state of the beryllium atom calculated in
two consecutive steps (see examples/be/be.lst and examples/be/be-1.lst).
../examples/be/be.data
../examples/be/be-1.data
- Hartree-Fock ground state energy of the hydrogen molecule (see
examples/h2/h2.lst).
../examples/h2/h2.data
- Hartree-Fock ground state of the BF molecule (see
examples/bf/bf_init2.lst).
../examples/bf/bf_init2.data
- HF calculations for the lowest
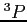 state of the carbon atom
(see examples/c/c.lst).
state of the carbon atom
(see examples/c/c.lst).
../examples/c/c.data
- HF calculations for the lowest
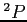 state of the
state of the 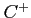 ion (see
examples/c+/c+.lst).
ion (see
examples/c+/c+.lst).
../examples/c+/c+.data
- HF calculations for the lowest state of the
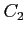 molecule (see
examples/c2/c2a.lst and examples/c2/c2b.lst).
molecule (see
examples/c2/c2a.lst and examples/c2/c2b.lst).
../examples/c2/c2a.data
../examples/c2/c2b.data
- HF calculations for the lowest state of the
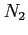 molecule (see
examples/n2/n2.lst).
molecule (see
examples/n2/n2.lst).
../examples/n2/n2.data
- HF calculations for the lowest state of the
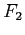 molecule (see
examples/f2/f2.lst).
molecule (see
examples/f2/f2.lst).
../examples/f2/f2.data
- A series of HF calculations for the the
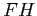 in external static
electric field.
in external static
electric field.
- no external field (see examples/fh/fh-0.lst)
../examples/fh/fh-0.data
- field strength -0.0001 a.u. (see examples/fh/fh-m1.lst)
../examples/fh/fh-m1.data
- field strength -0.0002 a.u. (see examples/fh/fh-m2.lst)
../examples/fh/fh-m2.data
- field strength +0.0001 a.u. (see examples/fh/fh-p1.lst)
../examples/fh/fh-p1.data
- field strength 0.0002 a.u. (see examples/fh/fh-p2.lst)
../examples/fh/fh-p2.data
- DFT calculations with LDA and LYP functionals (see
examples/dft/be-1.lst and examples/dft/be-2.lst).
../examples/dft/be-1.data
../examples/dft/be-2.data
- Two lowest states of the 2D hydrogenic harmonic potential (see
examples/oed/h3/h3-1.lst and examples/oed/h3/h3-2.lst).
../examples/oed/h3/h3-1.data
../examples/oed/h3/h3-2.data
- Lowest state of the Krammers-Henneberger potential (see
examples/oed/kh/kh.lst).
../examples/oed/kh/kh.data
- SCMC ground state calculations for the beryllium atom (see
examples/scmc/be-scmc.lst).
../examples/scmc/be-scmc.data
There are several standard names used by the program to keep track of its input and output
disk files. Normally the program writes out the data in the course of computations and
upon its completion into the following disk files:
- 2dhf_output.dat (a text (ASCII) file) containing the title of a case, the
time and date of its commencement, the number of mesh points, the internuclear distance,
the charges of nuclei and the number of orbitals, electrons and exchange potentials (see
writeDisk for details),
- 2dhf_output.orb (a binary file) containing molecular orbitals (in the
order specified by the input data following config label) followed by their
normalization factors, orbital energies, Lagrange multipliers and multipole moment
expansion coefficients (see write&sstarf#star;),
- 2dhf_output.coul (a binary file) containing
corresponding Coulomb potentials and
- 2dhf_output.exch (a binary file) containing all exchange potentials if the
exchio [in-one
 in-many] out-one card is present
in-many] out-one card is present
- fort.31, fort.32, ... (binary files) each containing the exchange
potential required for a particular pair of orbitals if the exchio in-many
[out-one
out-many] or exchio [in-one
in-many] out-many card is present
These files can be used to restart a given case or run another with slightly modified
parameters. If orbpot old card is present orbitals are retrieved from
2dhf_input.orb file, Coulomb potentials from 2dhf_input.coul and
exchange potentials from 2dhf_input.exch file (or fort.31, fort
32, ...files, if exchio in-many [out-one
out-many]). Note that there
is only one set of fort files.
In order to simplify the usage of the program, the xhf script (see tests/xhf) is provided
to facilitate handling of the disk files. The command xhf can be envoked with one, two or
three parameters. There are two basic modes of its usage:
- ./xhf file1 file2
runs x2dhf reading
input data from file1.data file and writing text data
describing the case into file2.dat file and binary data
with orbitals and potentials into file2.orb,
file2.coul and file2.exch files.
- ./xhf file1 fil2 file3
runs x2dhf
reading input data from file1.data and initial orbitals and
potentials from file2.dat, file2.orb,
file2.coul and file2.exch files and writing
resulting data into file3.dat, file3.orb,
file3.coul and file3.exch files.
If, for example, be.data file contains input data for
the beryllium atom (see Example 3) then
- ./xhf be be-1
starts and performs the first 300 scf iterations. Type
- ./xhf be-1 be-1 be-2
to continue calculations. In order to converge the SCF process
even better increase the convergence parameters (see the scf
label) and use the following
command
- ./xhf be-1 be-2 be-1
In addition, the xhf script can be used to perform the following
tasks:
- ./xhf stop
creats
stop_x2dhf file in a current directory to stop a running
program (see Section 5)
- ./xhf mkgauss filename
creates
symbolic links gaussian.out and gaussian.pun to
files filename.out and filename.pun,
respectively (see e.g. Example 5).
- ./xhf rmgauss
removes
gaussian.out and gaussian.pun files from a current
directory
- ./xhf clean
removes
*.[dat|orb|coul|exch] files
- The program should be easy to use provided you can start a calculation for a
specific system. You should not encounter any serious problems when the system contains
atoms from the first two rows of the Periodic Table. Then even the rough hydrogenic
estimates of the orbitals should prove adequate and after the initial couple of dozen of
iterations a smooth convergence should set in.
If, however, a system contains more than 15-20 electrons the initial estimates of the
orbitals have to be good enough to avoid divergences. Then, you have to choose the
parameters of the hydrogenic orbitals carefully or perform the finite basis set
calculations using the Gaussian94 to provide the initialization data for orbitals (see
Example 5).
One can also use HFS method to produce
initial estimates of orbitals and Coulomb potentials. For example, to start calculations
for the neon atom one can use the following input data:
../examples/ne/ne-hfs.data
This input produces good enough HFS initial estimates so that the calculations can be
continued at the HF level with the corresponding input:
../examples/ne/ne-hf.data
One can also use HF method with some model potential, e.g. the model potential of Green,
Sellin, Zachor (label POTGSZ).
- At the very beginning set the maximum number of scf iterations to something between
20 and 50 and/or impose crude convergence criteria for the orbital energy and
normalization.
- In case of convergence problems try to perform calculations on a sparser grid.
For example, the
![$ [61\times 79;30]$](img137.png) grid is sufficient to check the quality of the
initial data for the Ne
grid is sufficient to check the quality of the
initial data for the Ne system.
system.
- Choose smaller values of the relaxation parameters (
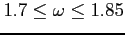 ) to
avoid divergences in the first few dozens of SCF iterations (rarely the values as small
as 1.2 may be needed). Subsequently the parameters should be increased to their (near)
optimum value (see the omega label and Example 8).
) to
avoid divergences in the first few dozens of SCF iterations (rarely the values as small
as 1.2 may be needed). Subsequently the parameters should be increased to their (near)
optimum value (see the omega label and Example 8).
It is possible to set
to its near-optimal value by calculating it from
a semiempirical formula; see the omega label. As a rule of thumb the optimal value of the orbital
relaxation parameter is somewhat smaller and, by default, is obtained by scaling the
value by 0.98 (see setDefaults).
- How to stop the program gracefully during a lengthy calculation
without killing the process and interrupting disk read/write operations?
All you have to do is to create a (zero length) file named stop_x2dhf in a working
directory by typing ./xhf stop (you can also use the Unix touch
command to this end). The program stops whenever this file is detected upon the
completion of a current orbital/potential relaxation.
This document was generated using the
LaTeX2HTML translator Version 2008 (1.71)
Copyright © 1993, 1994, 1995, 1996,
Nikos Drakos,
Computer Based Learning Unit, University of Leeds.
Copyright © 1997, 1998, 1999,
Ross Moore,
Mathematics Department, Macquarie University, Sydney.
The command line arguments were:
latex2html -split 0 users-guide.tex
The translation was initiated by on 2010-05-19
Footnotes
- ... program1
- J.Kobus and Ch. Froese
Fischer, Quasi-Relativistic Hartree-Fock program for
Atoms, to be published.
- ... time2
- The
maximum number of flags can be changed by adjusting the parameter
maxflags (see inputData).
- ...
system.3
- In this type of calculations convergence rates differ
greatly between orbitals. Therefore, if for a given orbital the
orbital energy threshold is reached it is being frozen.
- ... method4
- V.V.Karasiev
and E.V.Ludenia, Self-consistent multiplicative constant method
for the exchange energy in density functional theory, Phys. Rev. A
65 (2002) 062510.
- ... necessary.5
- A note of warning for the users
of the g77 compiler. You might encounter an I/O error when trying
to run cases requiring more than 70 exchange potentials. By
default g77 accepts file unit numbers in the range 0-99. If you
need more files to be opened you have to edit
f/runtime/libI77/fio.h in the g77 source tree, changing the line:
#define MXUNIT 100. Change the line so that the value of MXUNIT
is defined to be at least one greater than the maximum unit number
needed.
2010-05-19